Input box
The server takes as input a protein sequence or a list of sequences.
You can paste your sequence (or set of sequences up to a maximum of 50) selecting:
- Enter UniProt accessions (you need your Uniprot accession code/s)
- Paste your sequences (FASTA format)
- Upload a file (you may upload your file containing a maximum number of 50 sequences in FASTA format)
You can resume your work by inserting the Job-ID (anonymous) that is given you as soon as you upload your query. Alternatively you can provide an Email address to receive a notification when BAR+ ends the processing of your query.
BAR output
The output can be retrieved from an index page (Index table) collecting the summary of the results.
Results are retrieved in two steps: 1) sequences that are already
present in one of the 913.762 BAR+ clusters are returned; 2) all the sequences
that are found similar to sequences in the clusters are returned after a Blast search.
These are divided into sequences that meet the BAR+ scoring criteria to inherit all
the cluster features (Sequence Identity (SI) ≥ 40%; Coverage (COV) ≥ 90%); and sequences that can be associated to sequences already
in BAR+ clusters with SI < 40% and COV < 90%. According to our statistical validation
for these latter sequences annotation homology-based inheritance deserve some attention.
BAR+ output is provided in a table that contains the following information:
Cluster give a summary of the annotation provided by a
cluster as detailed below:

- Cluster identification
- Number of sequences in the cluster
- Average sequences length and its standard deviation (%)
- Sequence kingdom classification (Eukaryote, Prokaryote, Virus, unknown and other)

- GO terms in the cluster
- Fraction of validated GO terms
- All the validated GO terms. More granular GO terms are reported at the top of the list

- Number of total structures in the cluster
- List of PDB code/s
- Ligand ID list
- SCOP classification IDs of all the PDB chains in the cluster
- Total number of Pfam terms inside the cluster
- Validated domains from Pfam (when available)


Query all the queries submitted. When the cluster contains PDB/s, the templates and the query to template/s cluster HMM based alignment is provided.

- The query (in blue if there is a link to UniProt)
- PDB
- PDB position in the alignment
- Coverage (cov) and the sequence identity (id) of the template with the query
- The alignment in the PIR format suited for modelling with Modeller
Blast match provides some information about the match sequences. If the sequence is a UniProt entry the kingdom and the organism are provided, otherwise a brief description of the sequence as obtained from the source databases (Ensembl Genome Browser and RefSeq at NCBI) is given.
General Information
BAR is a web server for the functional annotation of protein sequences. The annotation process relies on a non-hierarchical clustering procedure of a BLAST all-against-all comparison genome wide comparison of 988 species and the entire UniProtKB (SwissProt + TrEMBL) without fragments sequences. A graph scheme is adopted in which each protein is a node. An edge is established between two nodes if the two corresponding sequences share a BLAST hit that undergoes the following constraints:
Coverage of the alignment ≥ 90%
The coverage is defined as the ratio of the length of the intersection of the aligned regions on the two sequences
and the overall length of the alignment (namely the sum of the lengths of the two sequences minus the intersection length).
Clusters are the connected components of the graph and are disjointed (each sequence belongs only to one cluster).
Statistical validation
The statistical evaluation of the GO and Pfam terms in each cluster is performed by computing the P-values as:
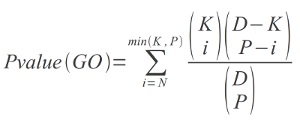
where D is the dimension of the database (total number of sequences which have at least one associated GO term),
P is the dimension of the cluster (total number of sequences of the cluster which have at least one associated GO term),
K is the number of sequences in the overall database with associated that specific GO term, N is the number of sequences
in the cluster which have associated the specific GO term.
Thus, the P-value is the probability of finding N or more proteins that have a given annotation by chance, given the
dimension of the cluster, the dimension of the database and the overall number of sequences in the database with that given
annotation. In our procedure a reliable P-value threshold for a molecular function GO specificity is chosen equal to 0.01.
Cluster annotation data format
This file is a simple text file with a double letter keyword at the beginning of each line followed
by a tab delimiter and another more specific keyword. If the character after the tab delimiter is a "#"
this means that the line is an explanatory comment for the following lines and can be skipped by a parser algorithm
(e.g. "FN #molecular_function_validated").
The meaning of the main fields is:
ID = the cluster name (an integer number)
DE = cluster description (size, average length, standard deviation, kingdoms)
FN = function field (GO terms)
DM = domain field (Pfam terms)
ST = structure field (PDB, SCOP, Ligands)
Description field - DE
The secondary keywords are:
SIZE = the number of sequences in the cluster
ALEN = the average length of the cluster sequences
DEVP = the standard deviation of the cluster sequences length
KING = the number of eukaryotic and prokaryotic sequences in the cluster
Function field - FN
For each line after the secondary keyword there are: the GO term accession,
the p-value, and the GO term definition (e.g. "FN BP_V GO:0006350 2.82565e-06 transcription")
.
The secondary keywords are:
MF_V = molecular function validated
MF_F = molecular function not validated
BP_V = biological process validated
BP_F = biological process not validated
CC_V = cellular component validated
CC_F = cellular component not validated
Domain field - DM
For each line after the secondary keyword there are: the Pfam term accession,
the p-value, and the Pfam family plus the Pfam description separated by a "#"
(e.g. "DM PF_V PF00046 1.68005e-23 Homeobox#Homeobox domain").
The secondary keywords are:
PF_V = Pfam domain validated
PF_F = Pfam domain not validated
Structure field - ST
The secondary keywords are:
PDB = pdb name, resolution in Amstrong, ligand list (when available)
SCOP = SCOP accession
LIG = ligand short name, ligand extended name , "|" separator , formula, "|" separator , molecular weight
(e.g. "ST PDB 2rdyA;2.03;MSE")
(e.g. "ST LIG MSE SELENOMETHIONINE | C5 H11 N O2 Se | 196.106")
(e.g. "ST SCOP a.4.1.1")